





Nat Commun | 潘东宁团队揭示甲基转移酶KMT5C非催化性新功能
2025年2月10日,复旦大学基础医学院代谢分子医学教育部重点实验室潘东宁团队在Nature Communications期刊发表了题为“Non-catalytic mechanisms of KMT5C regulating hepatic gluconeogenesis”的研究论文。该研究揭示组蛋白甲基转移酶KMT5C通过非催化机制调控肝糖异生的全新作用模式。

KMT5C是催化组蛋白H4K20me1生成H4K20me3的关键酶,其经典功能与基因沉默及异染色质维持相关。潘东宁团队曾于2020年揭示KMT5C在Trp53基因启动子区产生H4K20me3修饰,参与调节棕色脂肪细胞产热功能(PNAS, 2020, 117(36): 22413-22422)。此次,研究团队通过分析禁食与糖尿病模型小鼠的肝脏样本,发现KMT5C在饥饿和胰高血糖素刺激下显著上调,且其表达水平与2型糖尿病患者的空腹血糖呈正相关。通过构建肝细胞特异性敲除小鼠,研究人员发现敲除Kmt5c显著降低空腹血糖水平,并抑制关键糖异生基因(Pck1、G6pc)的表达。进一步实验表明,Kmt5c缺失导致PGC-1α蛋白稳定性下降,而外源补充PGC-1α可完全恢复糖异生功能,提示KMT5C通过调控PGC-1α稳定性发挥作用。
令人意外的是,体内外实验皆表明KMT5C甲基转移酶活性缺失突变体与野生型KMT5C具有类似的促进糖异生能力,提示KMT5C的甲基转移酶活性并非其调控糖异生的必要条件。机制研究显示,KMT5C通过直接结合PGC-1α的C端结构域,竞争性阻断E3泛素连接酶RNF34的结合,从而抑制PGC-1α的泛素化降解(见图1)。这一发现突破了传统表观酶“催化依赖”功能的认知框架,揭示了KMT5C影响PGC-1α蛋白稳定性的非经典功能。研究还解析了KMT5C自身的转录调控机制:胰高血糖素通过激活CREB1-CRTC2复合物,直接结合Kmt5c启动子并诱导其转录。这一发现将KMT5C纳入经典激素信号通路网络,为理解肝糖代谢调控的分子机制提供了新视角。
此外,在小鼠糖尿病模型(db/db小鼠及高脂饮食诱导肥胖小鼠)中,敲低肝细胞Kmt5c显著降低空腹血糖水平,并改善糖耐量与胰岛素耐量。临床数据分析进一步显示,2型糖尿病人群肝脏中KMT5C表达显著高于非糖尿病人群,且与空腹血糖水平正相关。这些结果提示,靶向KMT5C-PGC-1α互作可能成为糖尿病治疗的新策略。
综上所述,该研究揭示KMT5C通过非催化机制调控肝糖异生的新范式,突破了KMT5C表观修饰酶功能研究的传统框架。
复旦大学基础医学院潘东宁研究员、复旦大学附属中山医院青浦分院张敏主任医师为共同通讯作者,复旦大学基础医学院赵清雯博士(现就职于西湖大学医学院附属杭州市第一人民医院)为独立一作。该工作得到国家自然科学基金、科技部重点研发计划等支持。
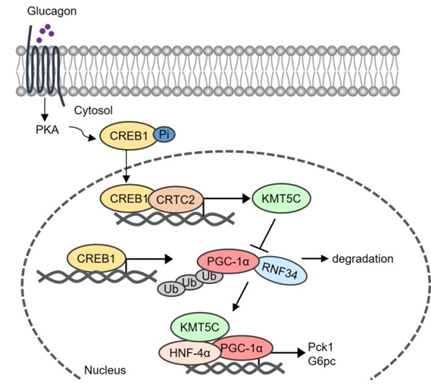
图1 KMT5C调控肝糖异生的模式图
原文链接
https://doi.org/10.1038/s41467-025-56696-y